潘锋:基于图论、大数据和人工智能的新能源材料基因探索
发布时间:2022-05-11来源:未知 编辑:admin
广告位置(首页一通--图文)
潘锋教授,北京大学讲席教授,博士生导师,北京大学深圳研究生院副院长和新材料学院创院院长。潘锋教授于1985年毕业于北京大学化学系,1988年在中科院福建物构所获得硕士学位,1994年在英国Strathclyde大学获得博士学位,并获得最佳博士论文奖,同年在瑞士ETH从事博士后研究。他长期致力于结构化学和材料基因的探索、电池和催化材料结构与性能及应用研究,发表了包括Nature,Nature Energy、Nature Nanotech、Joule、JACS 等内的SCI代表性论文300余篇。2020年任《结构化学》杂志的执行主编,获2021年获“中国电化学贡献奖”、2018年美国电化学学会电池科技奖与2016年国际电动车锂电池协会杰出研究奖。
以下是潘锋老师在材料人机器学习论坛上的报告文字整理版。观看报告回看请扫描文末海报二维码。
谢谢材料人的邀请,也感谢大家在周末时间来听我们北京大学深圳研究生院新材料学院团队在新能源材料的研究新范式方面的探索思考和进展。
>>> 材料基因工程

对传统的材料研究来说,大家都是从试错开始,一般是合成-检测-分析-再合成。现在大家都知道人工智能和大数据成为一个新的科学研究的新工具。但怎么更有效的运用大数据和人工智能?这里面我们需要一些新的数学方法有效地描述材料领域的结构与性能的关系。这些方法对我们材料领域来说是新的,但是这些数学方法不一定是新的,可能已经在物理、计算机和其它的很多的领域已经使用过很有效,具有强大的功能。
今天我向大家汇报的题目就是图论作为一种数学工具,可以用来构建高质量的材料大数据,有效地研究材料的构效关系。有了大数据以后,我们可以在数据里面应用人工智能去挖掘一些相关性,在新材料,特别是新能源材料的研发中,探索出“材料基因”,或者回答有没有材料基因,用来预测和决定材料的性能。这就是我今天报告的主要内容。具体内容分几个方面,首先跟大家探讨一下“什么是材料基因?” ,然后“如何开展材料基因的研究?”等。
说到材料基因,其名字沿用了是生命科学中“基因”,我们知道生命有基因,其决定了生命的上一代跟下一代之间的遗传。所以当我们在研究材料时,可以反复问材料有没有“基因”?材料基因的特点是什么?怎么研究?我们在做锂电池研究,可以通过探索锂电池材料的基因,来发现材料基因的共同特点,并运用于锂电池及其材料的研究。
先讲讲材料基因,材料基因这个概念由来是源于美国一些智库于2011年向奥巴马总统提出,美国未来的国家战略要引领科技,首先要在材料上引领,新材料研发与应用是基础。传统来说一种高性能新材料从发现开始到产业应用需要20多年,因此急需探索缩短这个时间,需要研究的新范式,目前已形成共识并成为各国科技竞争的发展战略。这里面我特别跟大家分享一下,什么是物质?什么是材料?如何从物质研究到材料开发与应用?物质是由其结构来定义的,物质结构(结构化学)是由相同或不同原子之间以特定的化学键连接(原子间分享电子)及其排序构成。物质是材料的基础,材料要针对不同需求对相应的物质进行调控。一个典型的例子是单晶硅,作为半导体芯片应用和太阳能电池应用的单晶硅杂质含量的要求是有巨大差别的。
美国2011年提出的材料基因组计划,试图改变我们刚才讲的传统材料研究的范式。我们国家也意识到通过材料研究范式的变革对加速新材料研发的重要性,科学院和工程院联合开了好多次研讨会,于2016年,我们国家设立了“材料基因工程国家重点研发计划”,材料基因工程就是说把生命基因工程的思想用到我们材料的研发中来,所以在第一期立了10来个项目,其中有一个项目就是关于材料基因工程来研发下一代全固态电池及其关键材料,我们联合高校和企业相关团队联合承担并完成了该项目。
什么是材料基因?
在承担项目过程中,我们不断问到底什么是“材料基因工程”,它的内涵及其外延是什么?这个命题虽然连在一起,实际上是两层含义:“材料基因”和“基因组工程”,前者是一个关于科学问题的定义,后者是关于如何开展的工程问题,工程就是方法和技术。可以参照生命基因工程成功的一些方法论。什么是材料基因呢?其关键是有没有“材料基因”,如果有它应该又是如何表达?我们可以先类比一下生命基因,然后再挖掘材料基因的关键要素。
>>> 类比:生命基因vs.材料基因
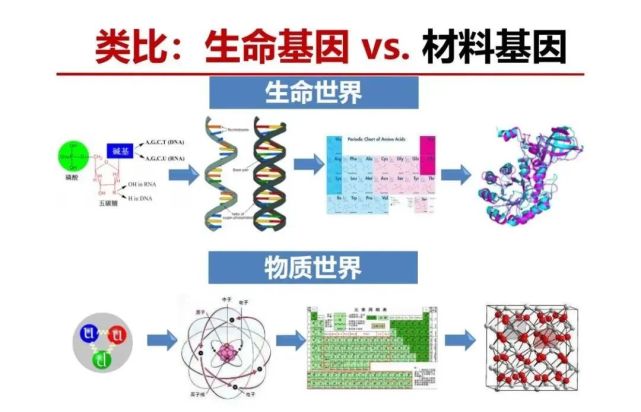
很久以来生物现象观察发现上一代和下一代某种特性相似,叫生命遗传现象,随着物理学和化学理论和测量技术的发展,才发现有一个决定生命遗传的“基因”,它有最基本的分子结构基,包括4种碱基的功能结构基元,碱基之间以共价化学键以特定的排序连接在一条链上,这两条链之间以氢键构成双螺旋结构,左边链和右边链之间象钥匙和锁一样以氢键为相互作用力以及特定空间耦合结构,是一一对应的,一旦知道了左边结构也就知道了右边结构,保证很精确的去复制。决定遗传性状就是一段这些功能结构基元的排序。因此生命基因里包含了三大要素,(1)基本的结构基元,包括碱基4种,相看起来比较简单但可以组合成非常复杂的排序,生命为什么选这种4种碱基,说实在我们现在也不得其解;(2)4种碱基的排序;(3)碱基之间的相互作用,包括链内是强相互作用的共价键(称之为化学键),链间是弱相互作用的氢键。
我们再看看我们的材料世界,什么叫材料?就是原子们在一起以不同的方式结合与相互作用。原子的特点是基于原子的质子数以及电子数,性质主要与外层电子数相关。门捷列夫从元素之间的性能相似性得到的周期表,可以说是第一张物质/材料基因规律的图表。量子力学的一大成绩能够发现和解释其原因,不同元素有不同的外层电子从而有不同的得/失电子的能力,这决定了它在构成材料时在结构基元中的角色与周围的排序与相互作用力。
所以,材料基因和生命基因有着相似的地方,从原子的尺度来理解生命基因和材料基因,二者关键共同要素都是功能结构基元及其它们之间相互作用和排序。一般认为一个材料特征和性能由其组成与结构决定,更深层次“材料基因”探索是由其功能结构基团以及相关的排序。
>>> 生命、材料vs基因

我们从宏观尺度来比较材料和生命,它们都应该遵循热力学定理,其演化都应该符合热力学第二定理,G=H-ST,G是吉布斯自由能,由能量项(H,焓,对应内能)与有序度项(S是熵,T是温度)来决定,系统是自由能是负的方向自发过程,当能量相在发生前后变化不大时,熵增加(无序度增加)会成为自发的过程。信息(需要输入能量)的本质是消除不确定性的量度,熵和信息对系统描述在本质上是一致的,是对物质世界和生命的信息表达。生命是一个有序的结构系统,在开放的体系里,能量的输入可以维持其有序运行和保持其有序度,避免不确定性因素的干扰其有有序运行。材料的合成或者材料的使用,实际上是在一个开放的体系里面进行,有一个能量流的输入可以驱动结构的序排,增加有序度。
>>> 生命与材料基因:三要素(结构基元、相互作用、排序)
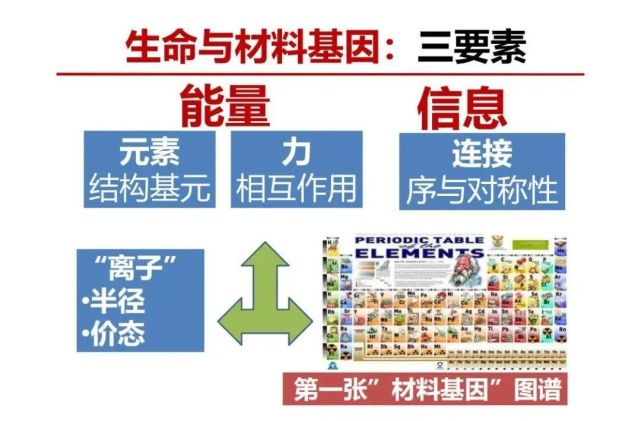
基于从微观和宏观尺度对比,用热力学分析材料和生命都有两大要素,一个是能量部分、一个是信息部分。能量部分,是物质之间的相互作用的强度,相互作用的强度由谁决定?肯定是由组成物质的元素,以及这些元素之间的得失电子能力的差异,叫电负性,形成不同互相作用类型,即不同化学键。当原子间的电负性差别很大,失去电子的形成正离子或者得到电子的形成负离子,正负离子之间形成离子键。当电负性差别不大,本身又不是那么容易失去和得到电子时一般是采用共享电子形成共价键。金属晶体中的金属共享游离的电子形成金属键。根据结构基元在材料中发挥的作用可以分类成为不同的功能结构基团。在锂电池中比较特殊的正极材料有过渡金属元素变价实现储能,过渡金属元素最大的特点是其外围d-轨道自旋电子,其氧化物的过渡金属配位多面体之间通过氧作为连接点形成d-自旋电子之间超交换相互作用。因此,材料基因的能量部分就是功能基团及其相互作用,有序度(信息部分)是它们的排序部分和对称性。
物理世界最有趣是它存在对称性,它贯穿在物理定理和材料的结构与性能,这可是成为我们研究材料基因的一个切入点。我们再看那个元素周期表,每个元素它在不同状态下,有其半径、价态以及与近邻的相互作用能力,开展材料基因研究,最好的办法是借鉴中国传统哲学‘道法自然’。自然界已经有很多自然存在的或已经人工合成晶体材料,可以向已知材料结构学习,因此需要把所有材料的物质结构收集起来形成材料大数据,然后挖掘材料基因的信息,开展基于数据驱动材料科学研究。
如何开展材料基因的研究?
>>> 材料研究新范式(数据驱动)

最近有一本书叫第四个范式,它把科学研究范式进行分类,第一个范式,是我们最早的人类进行了科学活动,开始制备科学仪器对自然现象进行观察,对观察的数据进行收集。例如开普勒等用科学仪器观察天体,积累了观察数据,得到地球绕太阳运动的数据。第二个范式是从这些数据找出科学规律,比如说地球围绕着太阳的运动是椭圆还是圆,还是圆很多的圆的叠加等等,最后牛顿总结出万有引力定律,自然界的运动规律以简单方程来描述,得到物质相互作用力(引力)都是跟距离平方成反比,天上的规律与地上的相互作用与相关运动的规律的一样的,由此建立了经典力学。在研究微观粒子相互作用的时候,发现了微观世界动量与位置不能同时精确测量的不确定性原理,形成了量子力学体系。爱因斯坦又发现了我们生活的物质与能量、空间与时间不能分割,可以统一来表达一个时空与物质能量的方程表达式,因此整个世界有了基础的相互作用的物理表达式。值得强调的是我们实际生活在的世界是复杂的,如海上的波浪以及鸟的飞行,都不能用简单的方程来描述,在做一架飞机等大工程时要用数据仿真和模拟,称之为第三个范式。现在我们已经有大量的科学和工程数据,复杂系统背后都有存在不同层次的相互作用,有一对一的相互作用,也有多体复杂的相互作用,这些相互作用背后是有底层的物理原因、逻辑关系,基于大数据和人工智能的科学与工程研发称之为第四个范式。
通过收集结构与性能数据,用数学表达和人工智能算法去挖掘构效关系,在材料科学与工程领域,开展基于材料大数据的结构和性能的相关性新范式研究。材料是个复杂系统,很难准确的计算,所以需要发现有效的数学表达和计算方法节省运算的时间,能够直插问题的本质,这是这个报告要汇报的一些思路和取得的进展。
>>> 材料大数据 vs 从数据到知识?

国外几个团队已经开展了材料的大数据建设,在锂电池领域伯克利Ceder教授建立了“Material-project”, 通过实验得到晶体结构数据计算出的能带和热力学参数以及锂电池的性能,很多人都在用。此外还有美国杜克大学和西北大学建设材料数据库,其包括了许多实验与计算预测的新材料数据。ICSD是传统的无机晶体结构数据库,它的来源是各国实验室发表文章的晶体结构数据。各国不同的材料协会,像美国陶瓷协会有自己的数据库,有机化合物有剑桥数据库等。
>>> 传统晶体结构化学:识别同构性挑战
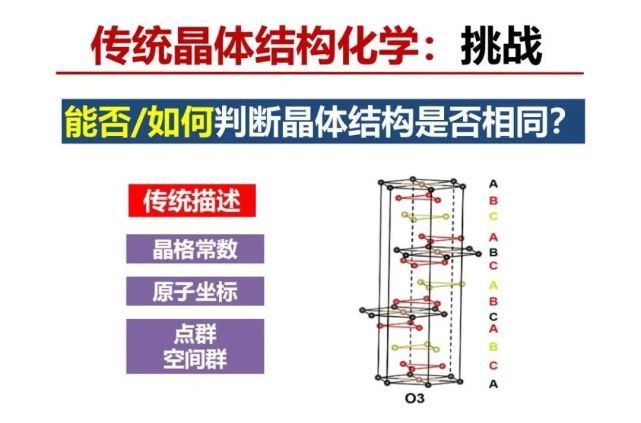
ICSD实验数据大概已经有18万个,每年都增加一两万个新的无机晶体结构数据,以氯化钠结构数据为例已经有34条,来自于不同年代和不同的实验室测量的数据,按照传统结晶学或结构化学的方法是用x-射线衍射来测得衍射点获得特定对称性以及对应的平行六边形晶胞格子类型(正方、长方、斜方、三角或六角等)、大小(晶格常数,a、b、c,斜方有面夹角α、β、γ等)、各原子在晶胞中有不同的位置,根据对称性可以得到多少个独立的原子位置,具有对称关系的原子可以用对称性换算出来。对称关系由点群中的对称操作(旋转、晶面、反演)描述,点群加上平移操作构成空间群。
>>> 传统结构化学挑战: 晶体结构的“测不准”原理
用传统的结构化学来看ICSD中的氯化钠立方晶格的晶胞参数,最小和最大差得非常大,约差0.04nm,比x-射线衍射精度(0.0001nm)大约多出两个数量级。不同实验室的制备条件的差异会导致材料存在缺陷不同以及测量本身有误差都会导致所测量的晶胞的原子坐标与间距存在差异。如果用测量单胞参数与位置来定义二个结构是否相同必然面临“同构性”判断的阈值设定的问题,即差异多大范围内才算同构的?会形成以下2种情况“不确定性”:测量误差阈值内认为是相同的结构也可能是不相同结构,以及测量误差阈值外认为是不同结构也可能是相同的结构。传统结构化学就有这么一个“同构性识别”的挑战,例如晶体结构中的晶胞参数a和b分别是 0.415纳米和0.414,如果误差的阈值设定在0.002,a和b两个之间没有差别,即a和b相同,如果阈值设定在0.0005,a和b是不同的。看氯化钠的ICSD数据,我们设定多大的阈值认为二个NaCl结构是相等的?SiC有250多种相结构,即使设定阈值很小,它也可能是不同的相结构。因此,对在设定误差阈值内认为相同的结构,会找到一个“黑天鹅”,是新的不同的结构。
我们在建立自己材料大数据的过程当中,面临的一个挑战就是用什么方法来表达或判断二个结构是相同(同构性)?测量总是有误差,要避免刚才讲的结构测量带来的“测不准”的问题,我们就想到另外一种思想,不去比较测量原子位置的绝对大小,而是大数据的思想研究事物之间的“关系”,即研究原子和原子之间的相互连接关系,把同构性判断转换成比较原子之间的连接关系,用连接关系的网络图来表达是否相同。
>>> 数学方法: 图论

数学图论是欧拉先创建,以此解决了著名七桥问题,把能否一次走过7个桥(不重复走)的问题抽象为点与边(两点之间的连通的表达成边,两点之间不连通就没有边)构成的图。点与点之间有连通与断开,例如两个点隔着河没有桥就不能连通。图论的基本思想就是说把是自然界的物可以抽象成“点”,点之间是连通可以用“线”表达,不连通就是断开的,这“点”与“线”就构成了一张图,我们把这个思想就用到晶体和材料中来表达结构。
>>> 结构化学新表达:图论

我们可以把晶体结构里面原子抽象成图论的点,如果这两个点有化学键可以认为电子在两个原子间是连通,表达成图论的线。什么叫化学键?就是两个原子之间有电子之间的交换,这就是化学键。如果一个原子把电子全部出,另一个原子把电子全部拿走,这两个原子形成离子键。如果两个原子的电子共享,那就是共价键,很多原子之间的结合都基于离子键和共价键,还有一种金属键是在全域范围彻底共享电子,大多数原子之间化学键是含有不同比例离子键、共价键与金属键的组合。因此原子间有化学键,就是联通的,没有化学键就不连通,从而构建化学和晶体的图论。
图论的点可以就研究对象来抽象。比如说我们在研究一个城市的特点的时候,我们可以把一个小区作为一个点,然后看看有多少条路跟这个小区和另外一个小区连接,我们也可以把小区把每栋楼作为一个点,然后再看到这楼和楼之间,楼到另外一个小区的楼之间,因此可以根据对象不同构建不同的图。同样在晶体里面不仅可以把原子当作点,也可以把原子组合构成功能结构基元当作点,研究其连接方式,形成不同层次的图表达。图的方式来表达最大的好处是可以用计算机里面的矩阵方程来进行运算,原子的实体空间位置转变成计算机虚拟的矩阵空间的数字进行数学运算的问题。
>>> 例如:图论 vs 同构识别
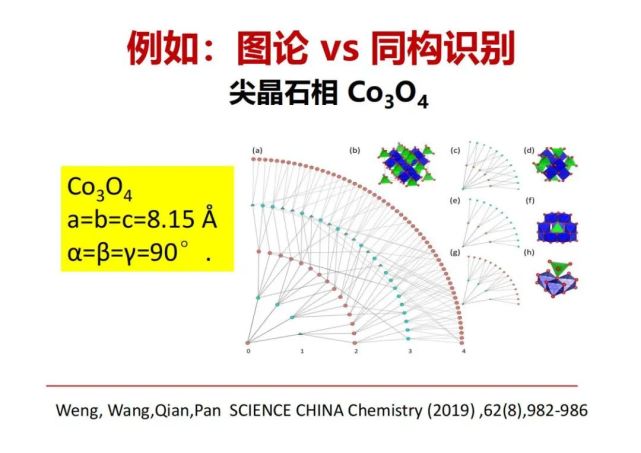
我们现在举一些例子给大家看看,这是我们发表的第一篇基于图论的结构化学的文章,用于解决刚才讲的晶体结构的同构性问题。以我们手机在用的锂电池正极材料钴酸锂的基础原料四氧化三钴为例,四氧化三钴在矿物学上叫尖结晶石相,一种氧密堆方式,立方晶系,晶胞长度都相同角度是90°,以一个晶胞为基础看有独立的几个氧几个钴,把每个钴与最近邻的化学键连接,再与次近邻的连接,再与第三层等不断地连接构成一张连接图。用这种方式可以发现实际上有三种独立的图,第一种以某1个钴为中心跟6个氧连接,还有1种是钴跟4个氧连接,还有1种以氧为中心与4个钴连接,这样就可以发现单胞里面有几个独立的原子构成独立的小图,这些独立的小图的组合构成尖晶石相的完整表达的大图。
>>> 例如:图论 vs 同构识别 :Li11Ti4Fe9O32 是否是尖晶石相?
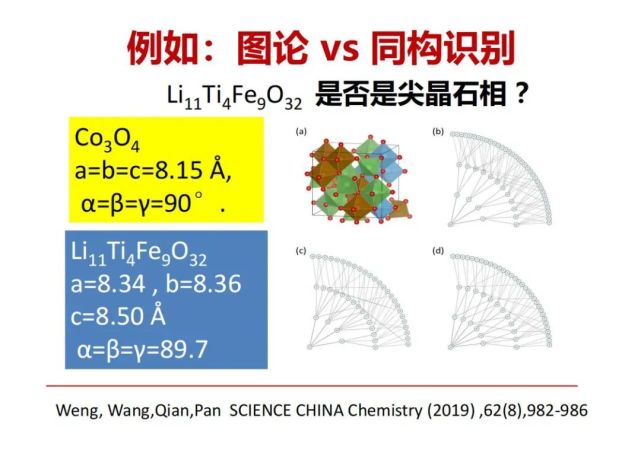
Li11Ti4Fe9O32像一个锂电池材料,它与尖晶石相Co3O4的晶胞特征不同(晶胞长度和角度都不相同),是最低对称性的单斜相,它是否是尖晶石相?用传统的结构化学方法很难识别。我们用以上图论的方法把单胞中的每个原子与最近邻、次近邻等4-5层构成连接图,发现实际上也只有三种独立,一种是金属与4个氧连接,另1种是金属与6个氧连接,还有1个是氧跟4个金属元素连接等。从图论的定义来看,这个材料锂、钛、铁、氧就是跟上面的四氧化三钴是同一种大图类型,因此可以确认它是尖晶石相。我们现在可以把传统的矿物学的每种晶体结构变成一种特定图的定义和表达。
>>> 图论 vs 结构化学大数据(北大)

基于以上所述基于图论的结构化学方法,发现刚才讲的氯化钠不同晶胞参数的连接方式是相同,从图论结构化学来说是相同的结构。用图论方法我们就可以把已发表所有的晶体结构拿来做数据清洗,也就是说用图论的办法来定义和识别晶体结构是否同构,得到了大概独立的约65万个独立的晶体结构,经过量子化学的结构优化计算,作为我们晶体结构大数据。
是否有化学键连接怎么判断呢?徐光宪先生写的《物质结构》的书后面有一个离子半径表(鲍林半径),根据结合原子两者电负性只差,可以初步判断是共价键还是离子键(是选用共价半径还是离子半径),如果是离子键,根据电价平衡计算是几价(对于可变价的原子)。它的原子半径,离子半径、金属半径来估算两个之间的距离,与实际原子间距离比较确定是否有化学键连接。因为如果原子间没有化学键,是范德瓦耳斯力,原子间的距离要大得多。用图论在识别的同构结构可以容忍有键角微小的差别,容许结构的一些畸变,这要连接关系一样可以作为同一类的图结构。
>>> 图论->代数图论
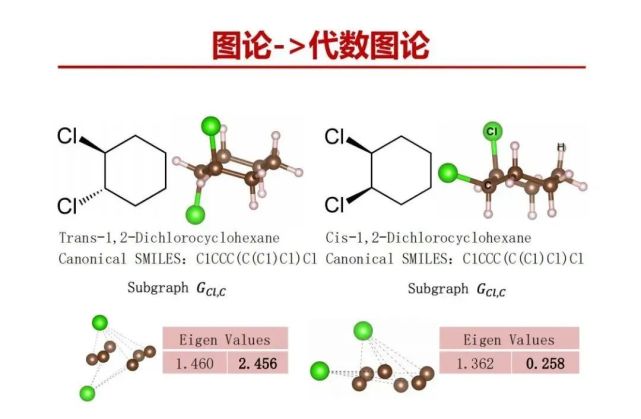
代数图论的持续同调是比较定量的图论方法,它能有效得到原子之间相互连接更细化的图信息。这个方法它选用了三种贝塔参数描述2点之间什么时候连接、3个点能否/什么时候连接在一起形成一个孔以及4个点能否/什么时候连接在一起形成一个洞。持续同调拿过程是两个相邻原子,以每个原子为中心,像波一样不断的径向扩大,到一定程度肯定会碰在一起,碰在一起就记录下碰在一起的空间距离,是两个原子之间的距离,用贝塔0表示。按照这个方式对三个原子进行关系进行描述,这3个原子沿着径向扩大碰在一起形成中间有孔,用贝塔1表示。按照这个方式对4个原子进行关系进行描述,有一种可能就形成一个洞,用贝塔2表示。代数图论的持续同调方法已经成功应用于生物医学领域,成功地开展了药物设计。我们把它用于材料研究。
>>> 用材料大数据研究:原子有效大小 鲍林离子半径的修正
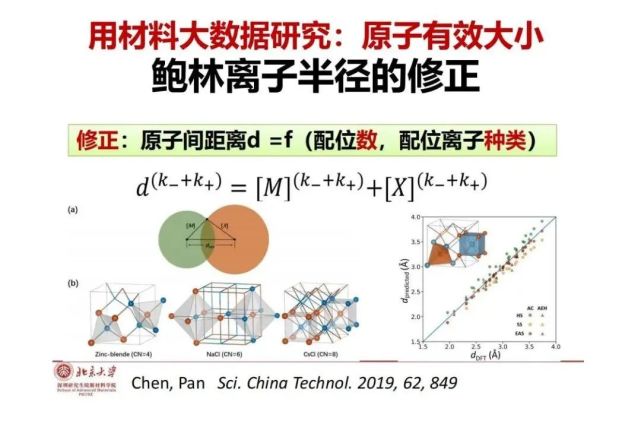
建立了大数据,我们可以检验一下《物质结构》书中鲍林半径表上的数据与不同实际晶体是否符合,以Fe为例,查到其鲍林半径与价态(Fe2+,Fe3+)和配位多面体(四面体,八面体)直接相关,通过这两项可以从离子半径的表格查到相应的半径值。如果我们把这些离子半径代入实际的晶体结构大数据中发现误差很大,原因是什么?我们发现在鲍林半径表格里面虽然考虑了价态和配位数,但没有考虑与谁配位,其使用的是“硬球模型”,但实际的中心离子和配位离子的极化性(让人家变形能力)和可极化性(自己变形的能力)是不同的,从而导致中心离子和配位离子(例如硫与氟的极化性和可极化性差别很大)会有不同程度的重叠,因为鲍林半径表没有考虑,所以就有很大的误差。如果我们把它考虑进去,用“软球模型”数学方法,从而加上配位种类对鲍林半径进行修正,可以更加准确的预测两个原子之间的距离。这是我们应用材料大数据以后的第一个工作,进一步探索了离子半径的真实的意义和在实际环境里面的大小的一种修正。
>>> 量子化学计算:晶体结构与电子结构的相关性研究(高通量和精确的电子结构计算 :HSE WKM)
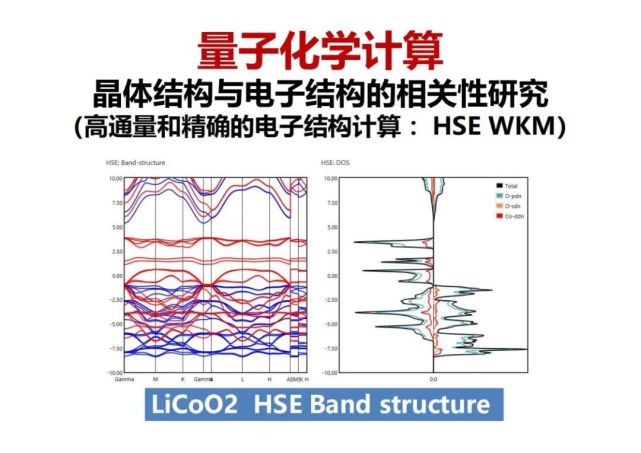
有了晶体结构大数据以后,运用人工智能的机器学习,希望把已有的一个晶体结构与电子结构关系学习到,然后有了新的晶体结构就可以预测电子结构。机器学习类似于人的学习,在识别一群动物是鸡还是鸭,先要通过了一些样本的训练,再放进一个鸡还是鸭,它马上就可以识别出来。类似有了一个晶体结构,用量子化学第一性原理计算,我们用了更精确的HSE方法,计算能带的带隙,再用机器学习来学习结构与带隙的相关性,然后再进行新结构的HSE带隙的精确计算用来修正机器学习,这样多次的迭代,得到90%以上的结构与带隙相关性(即一个结构就可以预测其带隙)的预测精度。
>>> 量子化学+人工智能: 机器学习建立力场

用量子化学和人工智能来研究原子间相互作用力,任何一个原子/离子放在晶体中,要感受到周围环境形成的力场。我们在固态物理中学习到简单的离子晶体相互作用,利用其周期性规律得到马德隆常数,运用该常数可以计算晶格能,其结果与实验测量很接近。对复杂系统的原子或离子迁移的动力学计算需要得到原子/分子相互作用的力场,有了精确的力场参数就可以快速的来预测物质的熔点、硬度和原子/离子的运动。
技术细节:用机器学习开发力场是基于势能面方法,选取Rcut范围内的原子作为近邻原子,近似认为中心原子只受近邻原子的影响。SANNP模型的特点是将DFT计算获得的total energy拆分到每个原子上,直接获得了每个原子的能量,传统的力场模型通常假设Etot等于体系中所有原子的原子能之和,拥有了原子能数据一方面提升了训练中包含的数据量,另一方面使得模型能更好的捕捉到表面等不同局部特征会带来的能量变化。力场方法的整个训练流程如下:1、提取近邻原子的局部环境特征。本文中使用两体势与三体势来提取原子间的相对位置关系作为局部环境特征。2、将中心原子的局部环境特征输入神经网络中,预测中心原子的原子能。右下图展示了我们的SANNP模型与DFT模型及经验嵌入原子势间的比较,可以看到SANNP与EAM相比较DFT的结果更为相似。我们用这方法进行了锂金属负极材料枝晶的生长模拟和预测。
>>> 大数据+人工智能: 预测结构与性质
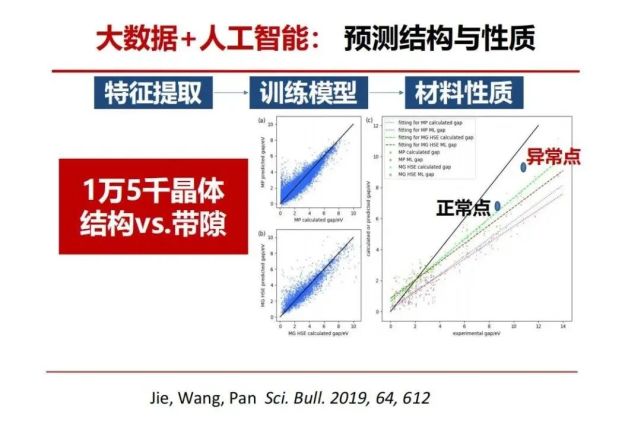
回到刚才将量子化学计算与机器学习相结合来从晶体结构预测电子结构,我们拿了15,000个晶体,进行量子化学计算与机器学习特征提取与训练相结合,进行反复的迭代改进训练模型,不断提高预测的精度。机器学习开展材料研究的工作现在越来越多,大家可能已经知道不少了,大家感兴趣的网上有许多资料。
我这里想讲的是人工智能和大数据还能干什么?在起初研究生命现象时发现植物的有些性状几代之间都有相似性,称之为遗传,分子生物学发现了遗传的本质是基因的复制。可由于遗传预测其相似的性状,但也发现有些时候偏离了预测称之为变异。我们在人工智能机器学习预测性能时总会发现一种现象,不管怎么改进的模型训练提高预测精度,即使预测已经做到百分之九十几了,总有那些不能被预测的部分,我们能否问这些是否是特殊变异的结构?我们把这偏离预测的(图中的“异常点”)材料结构拿出来看看是什么原因也可以看到变异的新物种。
>>> 大数据与人工智能:发现新型结构基元与电子结构
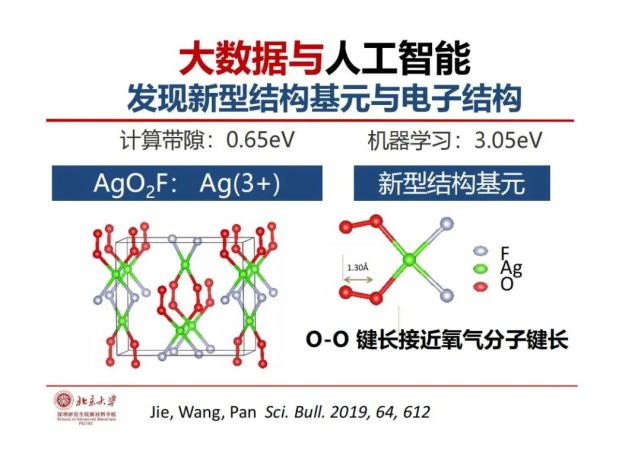
这里举了一个异常点结构的例子AgO2F,机器学习得到的带隙是3eV作用,量子化学计算的带隙只有0.6eV。进一步的结构分析发现银作为中心离子与两个氟和两个超氧离子配位,大家知道氧化性越大把电子拉过来的能力就越强,超氧离子和F都是强氧化性的。我们知道双氧水是氧化性很强的,最喜欢得到电子自己变成-2价,氟也有最强拉电子能力。银在这种环境下不是我们平常的+1价,而是+3价,超氧离子的能带在原来价带和导带之间,所以带隙就是不是平常预测的3eV,而是中间有超氧离子使得带隙只有0.6eV。
因此,运用大数据和人工智能既可以预测构效关系的规律同时也能发现不能被预测的异常点,从异常点中可以发现变异的新的功能结构基元—-新“物种”。
>>> 基于图论和材料大数据 发现新的2维、1维、0维材料
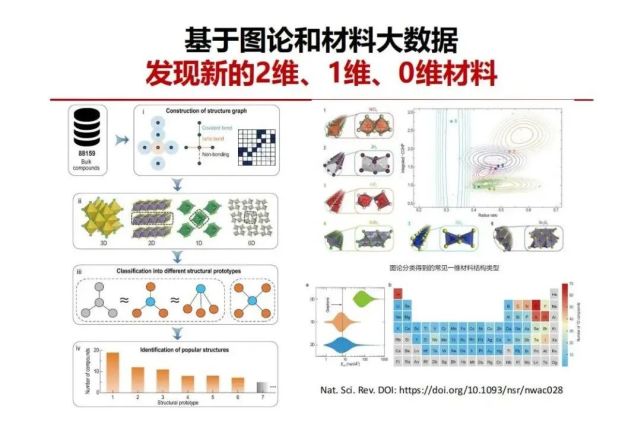
有了基于图论的结构化学和晶体结构大数据,可以重新来描述和定义什么是真正的低维材料,如2维,1维和0维材料:
二维材料就像石墨烯一样,可以用一个胶带纸不断地撕最后得到二维片,其在某一方向上没有化学键,原因是层之间的相互作用力是很弱的分子间范德华尔力。根据刚才图论结构化学,把在二维方向上有化学键连接成一个网络,但在与它垂直的方向上没有化学键,只有分子间弱的相互作用的材料定义为二维材料。以此类推,如果只有在一维方向上有化学键连接,另外两个维度都是没有化学键连接,只有范德华尔力,定义为一维材料。可以用胶带纸可以不断的撕,最后得到一维材料。同样如果他在所有维度上面都没有化学键连接,那就可以定义为零维材料。
运用图论低维材料的定义和结构大数据可以重新发现新型的二维、一维、零维材料,我们最近在国家科学评论上面发表了研究文章,技术细节:在材料的化学图论研究方法中,原子被定义为点,相邻原子间存在的化学键被抽象为边。我们基于88159种无分数占据情况的晶体材料,根据元素的电负性选择相应原子的共价、离子或金属半径从而确定原子间的近邻关系。通过该方法可以构造每种材料的结构图,并表达为邻接矩阵。通过图论方法,找到结构图中的独立联通单元,对其进行分析可获得该连通单元的维度,由此筛选出一系列二维、一维和零维(2D、1D和0D)材料。基于图同构比对方法,可以进一步将低维结构分成不同种类的拓扑结构类型。采用从第一性原理分子动力学方法评估一维化合物的动力学稳定性,从而排除不稳定的化合物。经过上述步骤后,总共得到了244种稳定的一维材料,这些化合物可被归类为138个拓扑结构类型。右上角图:为了进一步分析材料的化学信息,研究团队从成键特性与原子尺寸效应两个方面进行研究。其中成键特性由晶体轨道哈密顿布居(COHP)的积分项来代表,原子尺寸效应由阳离子中心配位多面体中正离子和负离子的半径比作为量化指标。一维材料在化学空间的分布情况反映出相同结构类型的材料拥有相似的性质,说明了图论分类方法潜藏着对材料性质的分类。右下角图:(a) 2D、1D和0D化合物的剥离能分布的类似小提琴形状图。灰色线表示由石墨烯的剥离能定义的阈值。接近半数的一维材料拥有较低的理论剥离能(胶带可以撕开),意味着这些材料均有可能在实验中被成功制备 (b)一维材料中元素组分的数目统计图,其中灰色的元素代表其不存在于任何一维化合物中。
由此我们发现了200多种新型的一维材料。一维材料有什么用?国际半导体组织规划的下一代半导体将把一维材料作为器件基础材料。目前用的硅基半导体,所有的硅单晶它都是硅的四面体共价连接成的晶体网络,当器件的沟道变得越来越小,到1nm只有十几个原子排列,边界将有10%左右断键,它还是硅吗?这些断键要界面重构形成钝化层,本征硅的性质将被调控,会因为硅的断键数不同及重构不均匀使得器件在原子尺度上的不一致而不稳定。所以真正的一维材料在自身方向是一维化学键连接,没有其它维的化学键要被切断和重构,从而在原子尺度保持稳定1维本征的性质。
这篇文章大家感兴趣,还可以看到一维和二维是怎么演化的,二维和一维之间相互转化的规律。还有一个有趣晶体化学问题,按照鲍林规则,两个金属配位多面体连接时以两个金属中心原子之间距离远(排斥力小)会比较稳定,两个多面体以共顶点连接最远,其次是共边连接,最近的是共面连接,大家想想如果两个金属元素越靠越近这两个原子的电子就会发生交互作用形成共享(Interaction),电子在多面体网络会形成渗流效应,这种电子渗流就使得材料具有半导体或导体的性质。
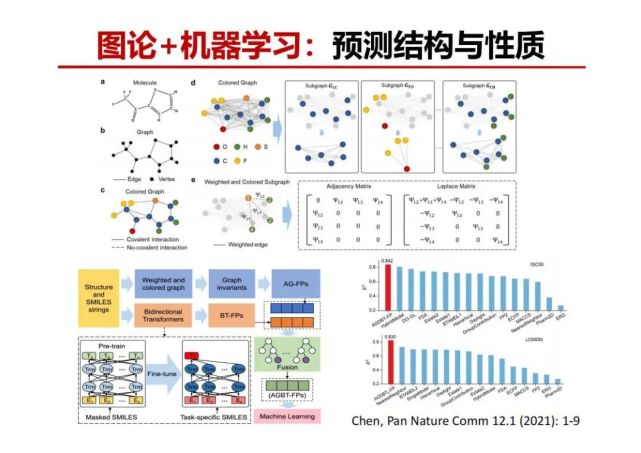
>>> 图论+机器学习:预测结构与性质
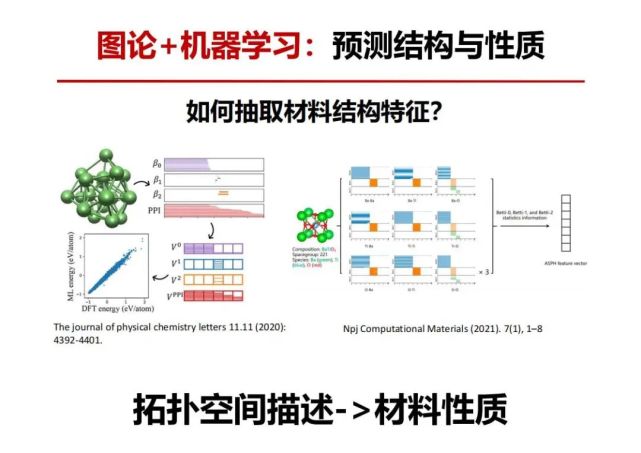
一个毫米大小的晶体有1020多个原子构成,10个纳米大小的晶体也有上万个原子,有趣的是这些晶体只有1种或少数几种确定周期性排列结构。例如碳原子构成的晶体常见只有石墨和金刚石。如果原子数少到只有几个或几十个时,原子堆积与排序的可能性会随原子数呈现指数级增加。这里面很有趣的问题是这种随着原子数目增加无限多的排序可能性是如何最后变成有限几种晶体?是如何过渡或演化的?我们如何去预测和发现?还有催化领域从纳米催化到目前很热的单原子或团簇(几个到几十个原子)催化都存在“原子数与其几何构型/电子结构及其性能的相关性”科学问题。
我们把量子化学计算、代数图论持续同调方法和机器学习相结合预测多原子堆积能量最低的可能结构,考虑晶胞中每个原子周围不同环境构成的持续同调,从而在拓扑不变量中引入原子信息。基于该特征所构建的晶体化合物机器学习模型,可以准确预测材料的形成能,误差仅为61 meV/atom。运用这原子间的短程作用和长程作用等数据信息,基于提取的拓扑特征,构建团簇能量预测机器学习模型。利用少量原子数团簇结构构成的数据集来训练所得的模型即可对中型和大型团簇形成非常高的预测精度。该模型可用于团簇结构的快速筛选,加快最稳定团簇结构搜索的速度。
>>> 图论+机器学习:预测结构与性质自监督学习->关键结构表示
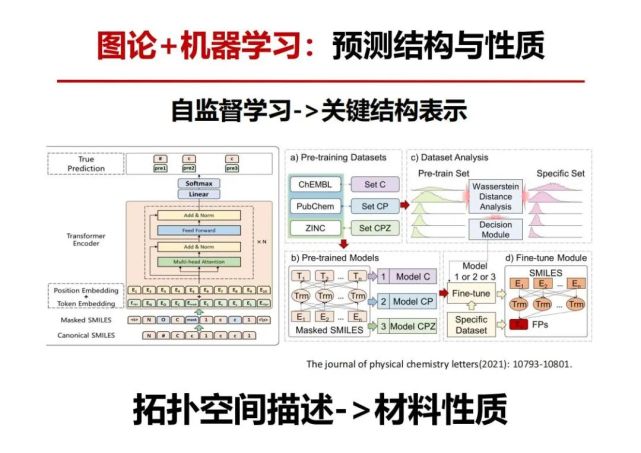
中国有一个关于矛和盾的故事,卖矛就是他的矛非常尖利,可以刺穿所有的东西,卖盾的说他的盾可以挡住世界所有的东西,那人们会问把能刺穿所有东西的矛去刺能挡住所有东西的盾的结果如何?自监督学习就是自己把自己的矛变得更尖利来刺穿自己的盾,盾要把自己变得更强硬不被自己矛刺穿,这样反复的改进和提升,自监督迭代学习。为了对材料的性质预测更加准确,我们运用自监督机器学习方法来预测结构和性质。
技术细节:左图利用“正确”的小分子字符串,即smiles进行构造,认为构造出“不完整“的字符串与相应的答案;然后在训练过程中,输入"不完整”的字符串,让模型推测出正确的部分。右图描述了文章的整体逻辑:基于自监督学习的策略,我们基于Chembl,ZINC,和PubChem三个大型数据集,训练出了三种预训练模型。并且基于这三个大模型,进行了一系列的下游机器学习任务(包含了回归任务,分类任务,以及virtual screening的任务)。
>>> 图论+机器学习:加入预学习用小样本数据实现准确预测结构与性质
有一次与大数据人工智能团队讨论,如何预测在一个机场里面判别一堆人是在一起讨论问题,还是在吵架及要引发打架?通过很多场景既学习“讨论场景”还要学习“吵架场景”,但这种感性的场景都是有限的。但人类关于“讨论”以及“吵架”有理性的概念定义与情景描述,如电影、小说、绘画等都有感性情景描述信息,对这些信息进行“预”学习,应该可以增加判断的准确性。其实我们人的学习也是不断从感性到理性,又回到实际的感性场景去判断与作决定。运用这个思想我们拿材料相关的知识作为“预学习”,使得对小样本材料数据预测的准确性大大提高。
我们在 Nat commun.讲了如何在有限的小样本数据进行机器学习实现高精度的预测有机化合物的性能,包括毒性等,有机物毒性样本数比较少(1万左右),但有机物多模态/维度的相关信息是大数据(SMILES 有机数据库有7亿个),基于大数据样本预学习,有效预测小样本有机化合物的毒性。
>>> 材料大数据反向设计
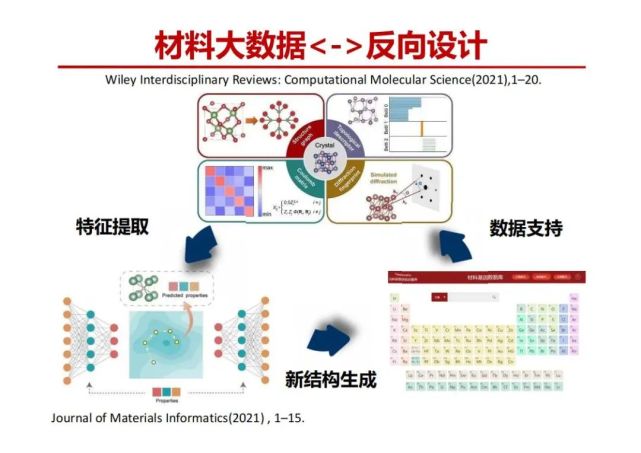
搞材料的人有一个梦想知道需具有特定性能的材料看如何把它设计和制备出来?这叫做反向设计,比如航天需要一个材料,需要硬度多少,韧性多少,一般就陶瓷很硬,金属有延展性很韧,容易变形,但它不容易断裂,能不能有一个材料又硬又韧,如何按照这个需求进行材料的反向设计? 我们对基于大数据和人工智能开展材料反向设计的研究进展作了综述,讲了反向设计的一个概念,即已有晶体结构数据库中的结构数据作为数据基础,通过材料特征提取方法提取结构特征,并将特征送入神经网络反向设计模型中反向设计出新的材料,通过DFT计算进行验证,将验证后的新材料作为数据库的补充,形成一个完整的数据迭代生成过程。
细节:左下图中展现的反向设计模型为VAE(变分自编码器),该模型由编码器,隐变量和解码器构成。变分的核心思想为强迫编码器从高维材料编码获得一个符合高斯分布的低维投影向量Z,即VAE希望将训练数据X的概率分布映射到高斯分布,使得人为输入任意一个高斯分布或者从材料空间按固定规则搜索到的隐变量,即可通过解码器获得一个接近真实的材料。同时在此过程中,通过将材料性质加入材料编码或者损失函数,还可以控制生成材料的性能。其大致的工作流程如下:1、通过卷积及多层全连接网络(漏斗状网络的左边部分)实现高维材料空间到低维空间(隐变量)的映射,该部分被称为编码器(encoder)。在VAE中,会对编码器添加约束,强迫该隐变量服从标准正态分布。2、通过另一个多层全连接网络及反卷积(漏斗状网络的右边部分)将低维向量投影回高维空间,实现对新结构的生成。该部分被称为解码器(decoder)。
>>> 知识图谱的诞生

讲大数据,我们看有些什么数据可以收集。刚才讲的晶体结构数据,这还是非常有限的,无机晶体结构已经合成的数据大概18万,加计算预测总共也就65万个独立。文本数据更大,我们已经有大概千万篇的科学文章发表,每年发表的文章的量不断在增加,人是读不过来的。还有非文本的照片(如电镜、x-射线照片)和视频信息。我们正在构建结构、文本、图片、视频等多模态信息融合,借鉴谷歌在2012年就提出了一个知识图谱,构建材料知识图谱来预测下一代材料。
许多人觉得谷歌的搜索比其他的搜索精准。大家知道要准确和快速找到目的地通常需要一种很好的地图。2012年,Google正式提出了“知识图谱(Knowledge Graph)”的概念,其初衷是为了优化搜索引擎返回的结果,增强用户搜索质量及体验。本质上,知识图谱是揭示实体之间关系的语义网络,以符号形式描述物理世界中的概念以及相互关系,实现准确而快速找到目标信息。
>>> 构建材料知识图谱
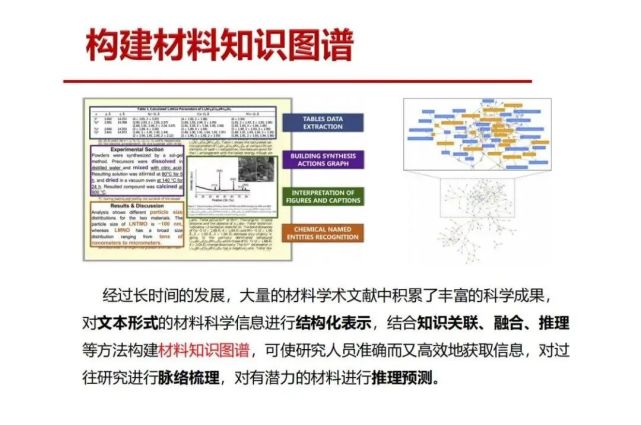
从Google “知识图谱”启发,我们构建材料知识图谱,把上面提到的多模态信息进行信息抽取出来。为了信息的融合,我们要对信息进行结构化处理,从而研究这些信息之间的关联,运用和发展数学方法看如何形成推理,构建一个材料知识图谱,形成对未来材料的推理和预测。
>>> 材料知识图谱整体构建思路

总的思路是材料学术文献信息提取需要对文本形式的材料科学信息进行结构化表示,结合知识关联、融合、推理等方法构建材料知识图谱,可使研究人员准确而又高效地获取信息,对过往研究进行脉络梳理,对有潜力的材料进行推理预测。要解决的一个关键问题是要准确地识别文献中的“主体(作者)+ 客体(材料)”。 材料是研究对象,是客体。是谁来研究?研究者是主体,一篇文章是包含主体和客体,及其相互关系,到底是“谁(主体)”在开展研究要搞正确,举个例子,北大有一个潘锋,清华有一个潘峰,英文都是Feng然后后面是Pan,或者是缩写F.Pan,这两个是否是同一个人(清华的F.Pan调到北大来了?)还是二个不同的人?
>>> 材料科学文献作者消歧—歧义化现象

科学文献里面要识别文章的作者是谁,几篇文章作者是不是同一个人,这与上面谈的两个晶体结构是否同构类似,是构建材料知识图谱大数据要解决的第一个问题,称之为“作者消歧”。机器学习与人的学习相似,“向好人会学好、向坏人会学坏”。机器学习也类似,学“坏”的数据(错误的信息)不会得出正确的判断,学“好”的数据(正确数据和信息)的就可能得到正确判断和结论。如果数据的错误多,得出错误的结论可能性高。避免作者信息搞错,先要进行作者消歧。
>>> 材料科学文献作者消歧—MatKG框架
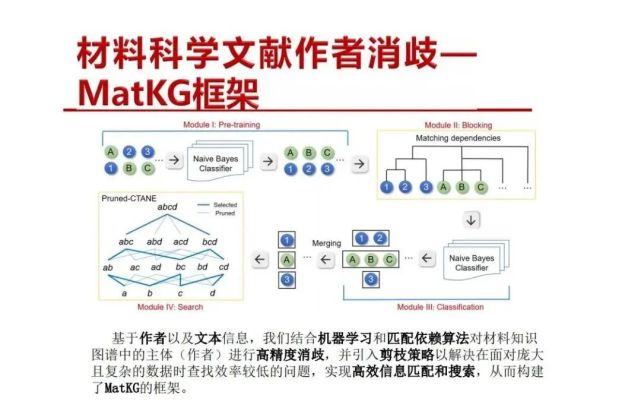
基于作者以及文本信息,我们结合机器学习和匹配依赖算法对材料知识图谱中的主体(作者)进行高精度消歧,并引入剪枝策略以解决在面对庞大且复杂的数据时查找效率较低的问题,实现高效信息匹配和搜索,从而构建了MatKG的框架。
2019年诺贝尔奖发给锂电池钴酸锂和磷酸铁锂的发明者Goodenough先生等。作为材料知识图谱的第一个应用,我们去复原磷酸铁锂这个材料发现的整个过程,诠释“前世”(第一次被发现,并不是作为锂电池材料用)和“今生”(作为锂电池正极材料被广泛应用)。在所有的过程当中,谁做了重要的Mile-stone的贡献。看到从发现作为锂电池正极材料到最近的广泛被应用经历了15年的时间。这之前不是作为锂电池材料的基础研究还要再追溯前10年,总共约25年时间。从中我们理解当一个材料从被发现/发明到被应用有着漫长路径,材料知识图谱可以让我们对于新材料的整个科学发现过程进行复盘,为了探索新材料提供借鉴。
>>> 材料关联与推理框架DATWEM—模型结构与表现
通过材料知识图谱能不能让机器把所有锂电池正极材料的文章读一遍,告诉我们下一代正极高性能的材料是什么?这是我们的初衷,相当于开始搞科学研究要去做调研,在做实验之前,要问自己为什么要这样做实验,实验做下去可能能发现什么新材料,这是“科学研究的注意力”。调研完会形成注意力,形成想法和推理。
>>> 材料关联与推理框架DATWEM——关联效果的显著提升
我们基于材料知识图谱双层注意力机制的多源数据融合,融入领域先验知识,显著增强可解释性,正极材料推导任务的效果全面提升。
>>> 材料关联与推理框架DATWEM——推理功能
DATWEM框架可明显改善正极材料的关联效果,纠正违
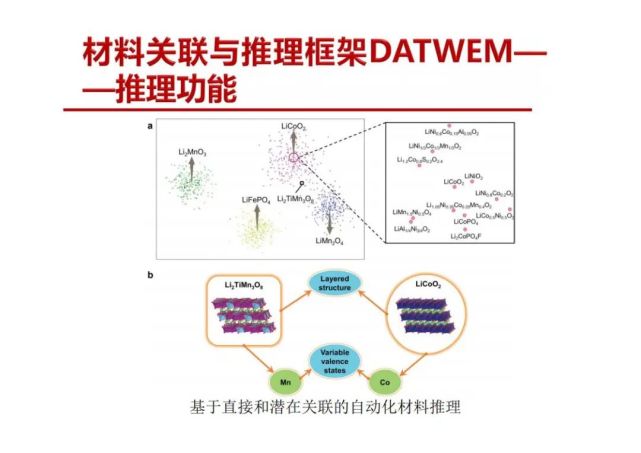
反材料科学知识的关联案例。通过自然语言处理技术构建数据驱动的材料信息探索的新范式,为新材料的发现与设计提供新见解。
>>> 文献大数据@构建材料知识图谱 预测高性能锂电池正极材料
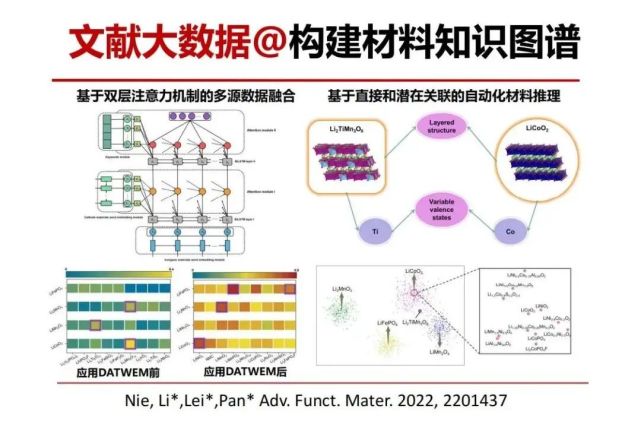
>>> 基于材料知识图谱预测Li2TiMn3O8作为潜在的高性能锂电池正极材料
用材料知识图谱相似的锂电池电极材料磷酸铁锂,钴酸锂,富锂锰氧,尖晶石锰酸锂等构建一种向量和它们之间的相似度。数学很奇妙,相似性的比较可以多维度。我们可以不用有线性的相似性比较,而是用非线性的余弦相似度,与统计概率相关,来预测下一代。双层注意力多元融合,去推理跟钴酸锂相似的是什么,然后跟其它正极材料什么相似,把各自的优点看能不能结合起来。这样预测到了Li2TiMn3O8材料,这个材料可能有潜在的应用,还需要用实验去验证。
技术细节:模型使用的机器学习框架为BiLSTM,LSTM即为长短期记忆(Long short-term memory, LSTM)是一种特殊的RNN,主要是为了解决长序列训练过程中的梯度消失和梯度爆炸问题。简单来说,就是相比普通的RNN,LSTM能够在更长的序列中有更好的表现。前向的LSTM与后向的LSTM结合成BiLSTM。传统LSTM无法编码从后到前的信息,举一个例子,“这个餐厅脏得不行,没有隔壁好”,这里的“不行”是对“脏”的程度的一种修饰,通过BiLSTM可以更好的捕捉双向的语义依赖。左上角图:在BiLSTM模型中融入双重注意力机制,融入领域先验知识(正极材料关键词),显著增强可解释性。左下角图:DATWEM框架可明显改善正极材料的关联效果,纠正违反材料科学知识的关联案例。右下角图:基于词向量降维后的无监督学习聚类正极材料图谱。输入嵌入量为62万个,保留了四种典型正极材料的大簇,以可视化不同材料之间的语义相似度。右上角图:Li2TiMn3O8与LiCoO2相似特征的自动识别。通过挖掘不同材料对应节点之间的直接或间接路径,可以从科学文献语料库中自动识别出与典型阴极相似度高的潜在正极材料。
>>> 如何系统提升新能源(锂电池)材料性能?
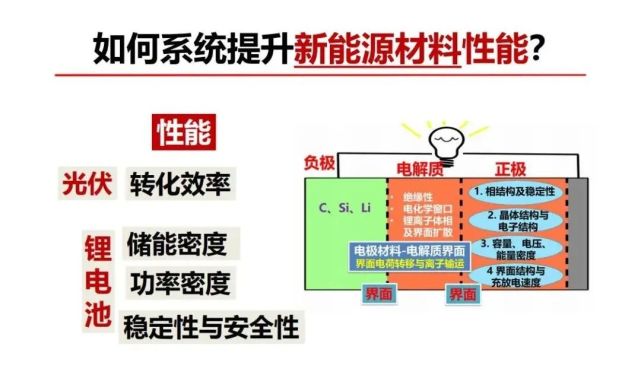
下面介绍如何把材料基因与新能源/锂电池材料要解决的科学问题关联起来研究。锂电池构架包含两个电极,实现外电路的电子流动和电池内部的锂离子穿梭,正极与负极它们之间有电势差,充电时外电路推动电子做功并把锂离子从正极推到负极,放电时电池对外电路做功,锂离子从负极被推到正极。电池性能最重要包括三个方面。第一个方面是储能密度,对应的是日常的手机能用多久,储能密度是锂电池材料存储锂离子的容量(能存多少个锂离子)和对应的电势差相乘的结果。锂离子存多了会互相静电排斥引发结构的稳定性下降和产生相变。第二方面是功率密度,对应的是充放电速度,经常听到锂电池充电时间太长太慢了,现在很多新的电池说可以快充,电动车和无人机加速要求快速放电,对应的是锂离子在电极材料体相中、电极界面处和在电解液中从正极到负极或反向的运动速度有多快。第三方面电极材料与界面的结构稳定性,对应的是使用循环性能与时间寿命。
>>> 研究思路:结构化学与材料基因从原子尺度揭示新能源材料结构与性能的相关性

关键问题;转化效率、储能密度 、功率密度、稳定性与安全性
>>> 结构化学方法:1. 创建基于图论结构化学/材料基因方法; 2.锂电池材料基因 与d电子结构化学;3. 构建界面结构/材料基因与电化学研究体系
让我们重新思考一下什么叫材料,材料是为了用的,用它的性能。要让晶体材料表现出优异的性能的必要和充分条件分别是其体相结构和界面结构。我们生命细胞要表现出功能,一定要经过它的细胞膜来实现。一个材料性能要表现出来,也一定要通过其界面来实现。材料界面与体相是有巨大差别的:第一,从对称性维度上面就有差别,晶体具有三维对称性,到了边界这种对称性不存在,结构基元的配位多面体对称性也破缺了,按照环境提供的条件要重新配位和重构界面。第二界面重构还有受到环境和使用条件的影响,电池要形成稳定的界面结构,是很难的一个问题,也可能由界面开始结构不断地被破坏,从而影响材料体相的结构稳定性。第三方面锂离子借助电解质当做桥梁让锂离子能够在正极与负极之间传输,现在使用的锂电池电解质都是液体,液体的特点是流动性和“润湿性”能够让锂离子在固态颗粒界面的空间距离最小(可以达到化学键的尺度)和传输面积最大化(覆盖整个颗粒)从而阻抗可以达到最小。固态电池的电解质是固体,会涉及到两个固体之间的接触问题,与液体不同硬的固体颗粒接触的空间距离大(达不到化学键的距离尺度)和面积小(只有固态颗粒接触的挤压应变承受的面积,属于点接触),因此电阻会很大,引发副反应、消耗能量,以及因为阻抗太大使得电极颗粒感受的有效电压会下降。所以固态电池要解决最大的问题就是接触阻抗。
现在锂电池负极是以石墨为主,为什么不用锂金属(锂金属的容量是石墨的十多倍)?如果要用的话需要解决哪些关键科学问题?锂电池领域获诺贝尔奖创新本质是开创了基于脱嵌机制锂电池及电极材料,正极相当于一个“旅馆C”,负极是“旅馆A”,正极和负极都为锂离子提供进出的存储“旅馆房间”,充电时锂离子从正极“旅馆C”自己“房间”出来到负极是“旅馆A” 自己的“房间”,放电锂离子又从负极回到正极,是定域的可逆的锂离子运动,很少会产生额外不可逆和不可控反应,体积变化小。可逆性(近似可逆)是锂电池长时间循环使用的物理基础。 锂金属作为负极的优点是容量高(4400mAh/g),但充放电的本质是不同的,伴随的是锂金属自由生长(非限域)沉积和溶解,引发锂枝晶的生长和一系列的“不可逆副反应”。
总结一下,锂电池的关键要素,1.正负极材料能存/放多少个离子,电压多高,构成储能密度;2.锂离子在正极里面传输出来,传输到界面,从界面又要传输到负极,这个速度有多快,称之为充电的功率密度,反向是放电的功率密度。3.充放电及在不同功率下的充放电的可逆性,这个与1和2点往往是矛盾的。 电池和材料如果设计得不好,容量太高和充放电太快(远离平衡状态)会引发相结构的演,结构可能产生不可逆破坏。此外,界面不稳定,会波及到晶体结构的相变,严重会引发安全性,充放电时结构的稳定性破坏容易释放出自由基,自由基会点燃有机电解液和相关的可燃物质,引发安全性问题。锂电池是类似生命的复杂的并有组织的体系,有一系列互相制约要素,这导致锂电池十几年来进步虽然有一点,但还是很慢的原因,所以我们用材料基因,大数据和原位大科学装置表征等方法来发现锂电池的相关相互结构制约的本质原因。从底层物质和机理的出发,即从原子尺度来发现结构和性能相关性,所对应的一门学科叫做结构化学(也有叫物质结构),结构化学就是研究物质在原子尺度排序和相互作用及其化学物理上所表现的性能。所以我们发展的图论方法表达它的结构,会更有效地探索材料的基因及其构效关系。
下面会举一个例子,在研究材料基因中,探索结构基元的相互作用会深入到 更深层次的过渡金属电子自旋电子相互作用。
>>> 新方法:基于图论的结构化学和材料基因
我们在2019年在《国家科学评论》定义锂电池材料基因:功能基元、作用力-键、连接-排序。
磷酸铁锂为例,为什么磷酸铁锂很安全呢?就是因为它有磷酸根,大家知道所有的阻燃物质你去查一下都是含磷的,为什么含磷?因为磷很容易捕获自由基。磷氧化合物和磷酸根很容易吸水,这两个要素是它阻燃原因。磷酸铁锂有磷氧共价键的磷酸根四面体、锂氧离子键八面体和过渡金属铁氧配位键(离子与共价键混合)八面体等三类结构基元。从结构基元来说有磷氧共价键稳定使得氧稳定,因此磷氧四面体起到了稳定结构框架的功能基元作用。锂氧八面体是传导锂离子的功能结构基元,铁氧八面体的铁从充电(锂拖出去时候)2价到3价及放电(锂嵌进去时候)的3价到2价,是储能功能基团。因此,锂电池电极材料一般可以解构成传输功能基元,储能功能基元、及构架功能基团。
现在电动车高能量密度的正极是三元高镍层状材料,据说特斯拉电动车大量用高容量的材料,大家已经发现其安全性不如磷酸铁锂,因为氧的结构基元是氧与过渡金属层3个过渡金属连接并与锂层的3个锂结合,以此氧只有配位键和离子键,没有稳定的共价键。
>>> 锂电池材料筛选:晶体结构与电子结构
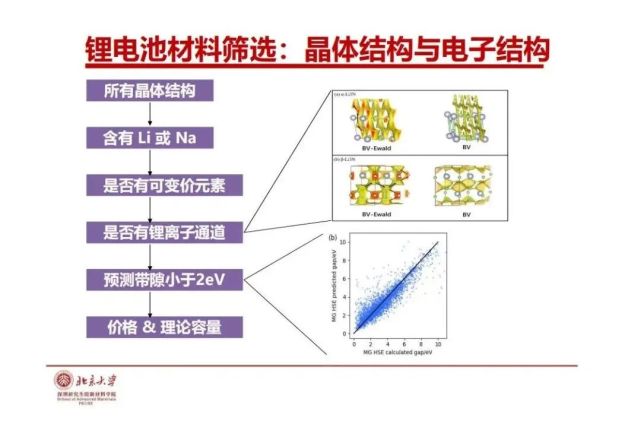
从晶体结构大数据我们可以筛选现有的锂电池和钠离子电极材料和固态电解质材料,作为电极材料它要两个关键要素,锂有可传输性,迁移能垒,低锂离子容易移动。我们发展了一种键价-Ewald(BV-Ewald)离子迁移模型,高通量发现结构大数据中哪些含锂或含钠晶体的Li或Na是“可迁移”的。此外,用于电池材料要求是电子能带结构的带隙不宽的半导体(导体更好,钴酸锂脱了一些锂成导体)。用于固态电解质材料要求对电子是绝缘的,电子能带结构的带隙要宽。我们可以用结构大数据按照离子可迁移性与电子能带结构的带宽去筛选可能的电极材料和固态电解质材料,发现新的结构基元、排序以及可能低成本的元素。
>>> 层状材料:结构基元与Li扩散路径
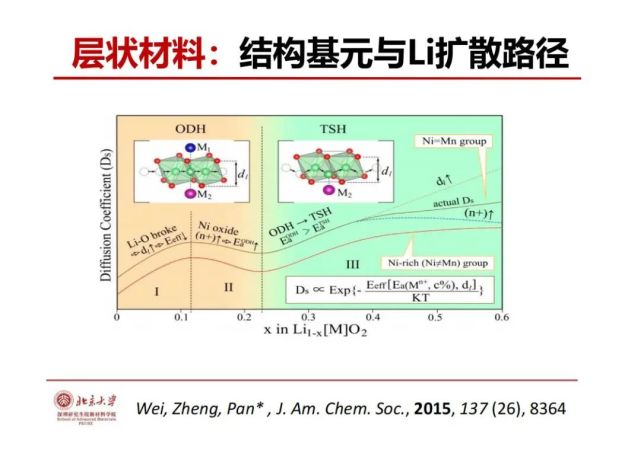
锂离子在镍钴锰3元层状(NCM)正极材料是怎么传输的?我们高端手机用的是层状钴酸锂而电动车许多用的是3元层状材料。用材料基因分析方法,层状材料的锂层由锂氧八面体,锂离子满的时候锂离子传输从一个八面体传到邻近一个八面体(八面体-八面体传输),受到两边过渡金属的相互作用。当锂跑了20%左右,锂离字倾向于从八面体到四面体再到八面体(八面体-四面体-八面体)的传输路径机制,当层间距大对应八面体也大和锂氧键也长,氧对锂的束缚弱,锂离子传输的能垒会低从而传输会快,这是结构基元与锂离子传输相关性的一个关键要素。
>>> 层状材料:中子测量原位全电池Li扩散动力学
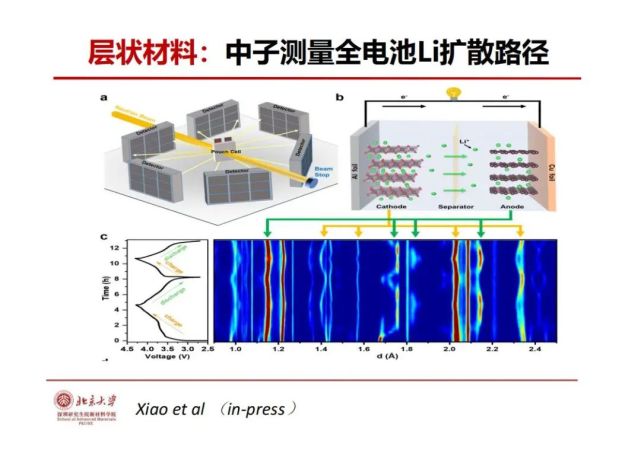
2015年我们理论预测层状3元锂离子的传输机理。现在我们可以用国家(东莞)散裂中子源的衍射谱仪原位检测实际锂电池原位锂离子在不同的电压下的传输动力学路径,与理论的预测基本一致,新的中子表征观察发现是当锂脱出40%以上时,锂是八面体-八面体以及八面体-四面体-八面体二种传输机制同时存在。
>>> 锂离子高电导:结构基元与协同传输
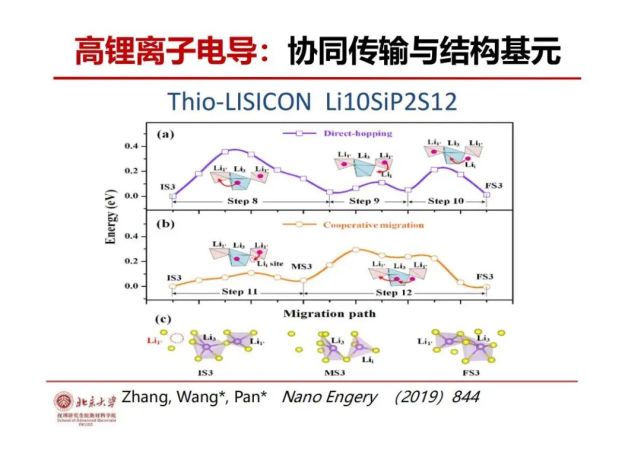
怎么让锂离子在固体里面能够快速传输?甚至传得比电解液更快?最近实验发现在Li-P-S固态电解质比液体锂传输要快,用材料结构基元及其它的连接方式来研究其锂离子传输机理,发现其快速传输的机理是相邻的锂离子结构基元之间有互动,就相当于构成一个协同传输的超结构基元,一个低能垒结构基元(称A)的锂离子传输的过程中会影响相邻结构基元(称B)的构型从而降低了其传输能垒加速B的锂离子移动和传输,B的锂离子移动反过来又影起A的构型进一步变化使得其能垒更低传输更快,形成了正反馈互加速锂离子的协同传输。
>>> 三元层状锂电池正极(NMC)热稳定性vs 结构基元
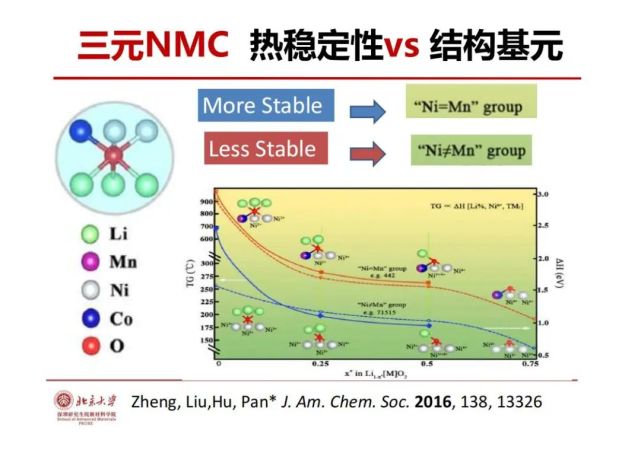
锂电池怎么做得安全和稳定?从材料基因的角度去挖掘结构的稳定性,剖析一下锂电池的充放电过程,对层状的材料来说,最重要安全性和稳定性是氧的稳定性。氧的结构基元是氧与过渡金属层3个过过渡金属连接并与锂层的3个锂结合,以此氧只有与过渡金属配位键和与锂的离子键,没有稳定的共价键。充电的时候把锂拖出去,氧上的Li从三个锂变成两个、再变成一个直到全部脱完。用热分析发现其热稳定性从900℃一直会降到150℃左右,层状材料的结构稳定性与锂有没有在锂层密切相关,即锂层是空的及Ni4+会导致氧的稳定性下降。
>>> 构效关系:锂电池材料d电子结构化学
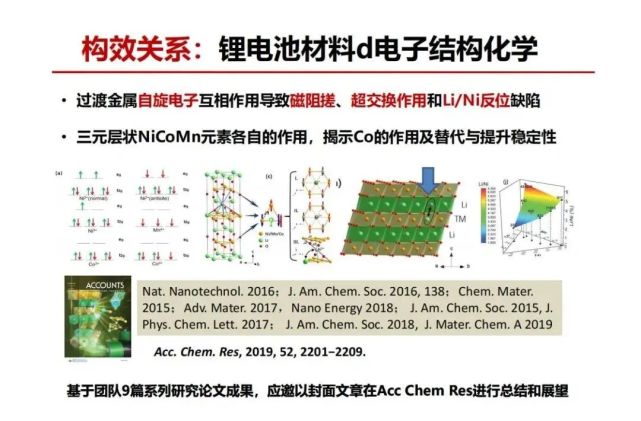
怎么能够使得层状的氧的结构更稳定? 在NMC三元材料总有锂跑到过渡金属层同时镍跑到锂层来称之为Li/Ni反位 ,还有过渡金属是可变价,不同价有不同的自旋电子,比如说镍二价在八面体有两个自旋,三价是一个自旋,不同的自旋电子互相作用会导致什么结果?Goodenough教授早年他还没有做锂电池的时候,发现自旋电子相互作用一些规律。什么叫化学键?自旋向上电子与自旋向下的电子配对,变成一个单键。如果过渡金属通过中间一个氧连接,d-过渡自旋电子之间会形成超交换相互作用,如果3个过渡金属通过氧为桥梁形成三角的连接关系,3个过渡金属只有2个有自旋超交换作用第3个没有自旋电子,这两个配对是稳定。如果3个都有自旋,这是“三体问题”是不稳定的,它们之间的自旋相互作用就会干扰,这干扰叫磁阻搓,使得原子间的结合力变弱,如果第3个没有自旋电子,那就没有磁阻搓,结合力就强。用3体自旋电子相互作用与磁阻搓分析,Li/Ni反位使得锂到过渡金属六边形中间替换成没有自旋的锂可以消除磁阻搓。钴三价没有d-自旋电子也能消除磁阻搓。可以预见Li/Ni反位量与过渡金属层的自旋电子数有关,Co3+没有自旋,含量越多Li/Ni反位越少,Mn4+有3个自旋,Mn4+越多Li/Ni反位越多,我们用中子衍射测量了自旋电子数量与反位数量相互关系,得到相关的相图,证实了我们的预见。
总之,我们系统研究过渡金属自旋电子互相作用导致磁阻搓、超交换作用和Li/Ni反位缺陷vs.三元层状NiCoMn元素各自的作用,揭示Co的作用及替代可能性。基于团队9篇系列研究论文成果,应邀以封面文章在Acc Chem Res进行总结和展望,大家感兴趣可以再看一下。
>>> 三元NMC 热稳定性vs 结构基元
Li/Ni反位使得锂层Ni2+与过渡金属层的过渡金属之间有180度的较强的超交换相互作用,有约60meV(室温是25meV)远高于室温的稳定性,该反位在锂层的镍氧八面体成为了稳定结构框架的功能结构基元,同时氧的结构基元也更加稳定了。因此,Li/Ni反位缺陷能增强NMC三元层状的稳定性。
>>> 发现高电压高容量钴酸锂的机理
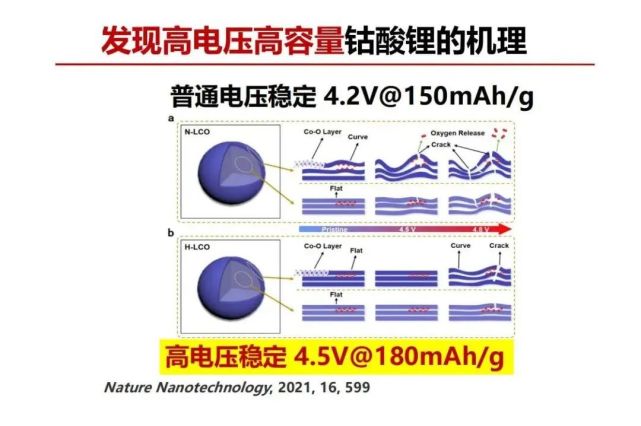
现在高端手机用的正极材料是钴酸锂。自从Goodenough教授发现后用到锂电池正极大概十几年,容量稳定应用只能发挥近理论容量(280mAh/g)的一半,即145 mAh/g电压在 4.2V,超过这个容量和电压就会出现不稳定,其原因一直不清楚。近几年发展成功容量180 mAh/g其电压可以提高到4.5V,有几篇文章对其原因讲得也不太清楚。大家可以去比较几篇发表文章中4.2V稳定和高电压稳定4.5V钴酸锂材料的多晶x-射线衍射(XRD)谱,看不出什么差别。让我们问多晶XRD测量方法的本质是什么?它是把多晶的每个颗粒的晶格周期性的衍射信号叠加得平均值,测得衍射方向(hkl)的间距(d-值)是平均值,并不能反映每个颗粒以及一个颗粒的不同位置的原子排列信息。因此,要发现普通钴酸锂和高电压钴酸锂的差别一定是要测量微观结构差别,这需要研究单个晶体颗粒以及一个单晶颗粒不同位置(从界面到晶体中心)的微观结构。我们与北大化学院孙俊良老师一起用自主改进的3D电子衍射的技术,将透射电镜的电子束作为衍射粒子/波的源,透过微米大小的钴酸锂单晶(透射厚度要在约1-2微米左右或之下),可以选单晶的不同部位来检测。这个检测技术可以看到钴酸锂不同电压充放电前后的微观结构变化。锂电池材料结构检测要包括本征结构以及使用过程的结构变化(包括其结构稳定性和变化的可逆性),电池材料在电化学环境下充放电,电子和离子从界面进/出,因此结构的破坏往往会从界面开始。我们用这技术看到了普通和高电压稳定钴酸锂的差别,普通层状钴酸锂的层有不同程度弯曲,在充电(锂离子脱出,层间距扩大)过程当弯曲会放大,前面讲过层间距大锂离子迁移容易,弯曲层状结构的层间距是不一样大的,层间距大锂离子先脱出,一旦锂离子脱出在该处的层间距就会进一步扩大,这样就会形成一个正反馈导致层的弯曲的不断扩大,弯曲处形成的应力越来越大,当应力积聚到一定程度就可能导致层的断裂,过渡金属会发生迁移同时氧原子就有可能释放出去。相对比的是用该方法检测发现高电压钴酸锂的层是比较平整的,按照以上讨论类推在充电时平的层的层间距是比较均匀的扩大,产生应力的积聚就小,层能保持较高的稳定性,因此能承受高电压下高容量锂离子的脱与嵌。
锂电池层状材料可以想象成一块块木板平堆起来,堆积木板的中间因紧密接触是平整的,但堆积木板的边往往会翘起来不易平整,要把它搞平整,在建筑上是在边上钉钉子来防止它不平或减少弯曲(翘起来)。层状钴酸锂的高电压的原理与木板堆积相似,要经得起环境的干扰使得堆积从中心到边缘始终保持一致,因此,其解决方案是在边缘“钉钉子”。
这个工作就给大家一个启发和进一步思考什么是物质,什么是材料的基本问题。材料的目标是使用,是使用其性能,这与材料从中心到界面的结构一致性密切相关,特别是与界面的性能有直接的关系的材料。
>>> 发现高性能锂电池无Co层状正极材料
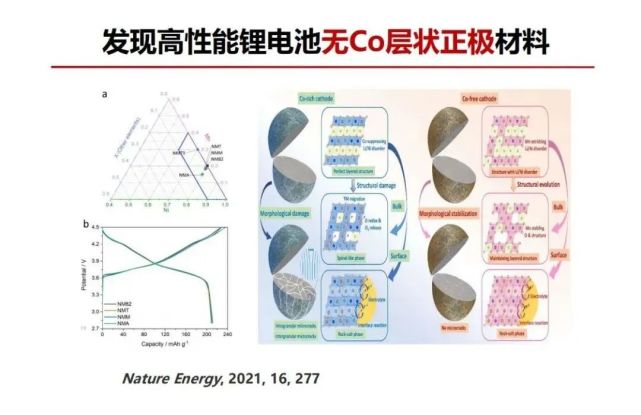
回到材料基因,结构基元的最小的单元是元素特征,即其原子或离子的外层电子数,在锂电池层状正极材料的过渡金属的不同价态有不同的d-自旋电子,钴氧八面体中Co3+是没有自旋的,钴是战略物资很贵,3元中能不能不要钴,材料通过Li/Ni反位又可以更加稳定,过渡利用金属层镍与反位到锂层的镍之间就可以形成180度超交换相互作用把材料变得更稳定。基于这个思想我们设计多元层状材料没有钴,实验证明可以实现无钴正极材料并在高电压下结构更加稳定,开启无钴层状正极材料新材料研发。特斯拉 CEO也讲,对大规模使用电动车动力电池,不仅钴即使镍也太贵了,因此连镍都要减少,这是我们正在研究的一大方向。
>>> 界面材料基因vs.电化学体系推动发现界面有序水结构

刚才讲到界面是非常重要,尤其是电化学体系,例如锂离子在电池界面怎么进去又怎么出来?在电池和电催化的环境下,所有的离子都是带电的,带电离子会溶剂化,如金属阳离子要与有机溶剂和水形成溶剂合多面体,电极静电的作用下在其界面附近会变成有序排列或脱嵌。我们这篇工作与厦大李剑锋老师合作,在研究电解水制备氢气,发现水在界面上有两种结构的水,一种是溶剂水其本身是由氢键构成的网络结构,另一种是4个溶剂水与阳离子配位的溶剂化水,在负电位下的负极与水合阳离子静电相互作用,把溶剂化的水有效地拉倒电极界面形成有序的排列,并且使得水上带正电质子与负极的有效距离缩短,形成电子-质子有效的电荷转移,实现更有效地水分解产生氢气。总之,原子尺度原位发现电解质溶剂在电极界面的有序结构,对制氢/燃料电池和高功率锂电池有重要指导意义。
>>> 新范式@新材料
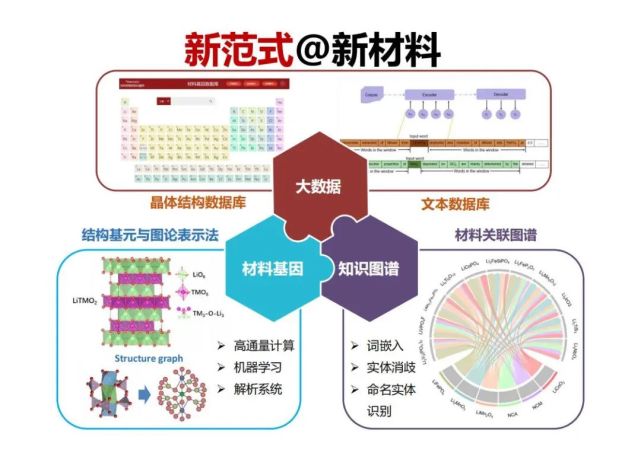
下面总结一下材料研究的新范式,融合大数据、材料基因、知识图谱可以加速新材料的研究与开发,通过构建锂电池基因大数据是加速高性能材料研发的新范式 。
>>> 基于图论和材料大数据 创建晶体结构解析系统
还有一个事情跟大家汇报一下,现在有些国家对我们实行科技软件的禁运,包括两三个物质结构分析软件,这些分析软件的功能是从材料的x射线衍射数据模拟和判断出物相、解出基本结构和精修结构存在的缺陷,这些结构分析软件用的是我们上面讲科学范式的第三个范式—基于模拟和计算。
>>> 材料基因大数据@ 结构解析(新范式解决晶体结构软件卡脖子的挑战)
>>> 展望:图论@结构化学 vs 新材料
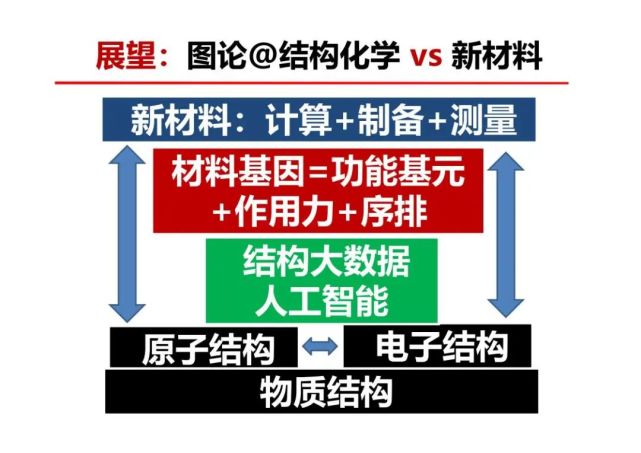
加速材料研究,可以同时进行正向(自下而上)方法,从底层的物质结构(原子结构与电子结构)、结构大数据结合人工智能算法、由结构基元及其排序与相互作用的材料基因到新材料的设计与计算及其制备与表征。同时可以进行逆向设计,从需求出发来设计材料,寻找适合的功能结构基元与相关的元素及其它们的排序和相互作用。然后进行可控制备和测量结构与性能,看是否与预想的一致,特别是运用同步辐射x射线和中子大科学装置进行原位与非原位的高精度测量材料制备及其应用过程中的结构演化。总之,这种基于图论结构化学和材料大数据、大科学装置等开展正向和逆向的预测与实验的循环迭代,是加速新材料从基础研究和产业应用的新范式。